Eph4 drug repurposing (Ligand-Protein analysis)
In this tutorial, we will reproduce the results of Gu S. et al. 2018 (https://www.nature.com/articles/s41598-018-25790-1). In their work, they searched a dataset of FDA-approved drugs for inhibitors of the receptor tyrosine kinase erythropoietin-producing hepatocellular A4 (EphA4). This receptor was identified as a molecular target for Alzheimer’s disease (AD). They selected and tested 22 molecules and found 5 potential inhibitors of EphA4. Specifically, nilotinib (https://go.drugbank.com/drugs/DB04868), a kinase inhibitor, inhibited the binding of EphA4 and ephrin-A at a micromolar scale in a dose-dependent manner. In this tutorial, we are going to use a structure of EphA4 (PDB code: 2wo2, https://www.rcsb.org/structure/2WO2), the same structure Gu S. et al. used in their work.
Preparing the Target
Open PyMOL and run the following command to download the structure of EphA4 as well as remove EPHRIN-B2 chain and water molecules to prepare the structure for docking:
fetch 2wo2 remove chain B remove solvent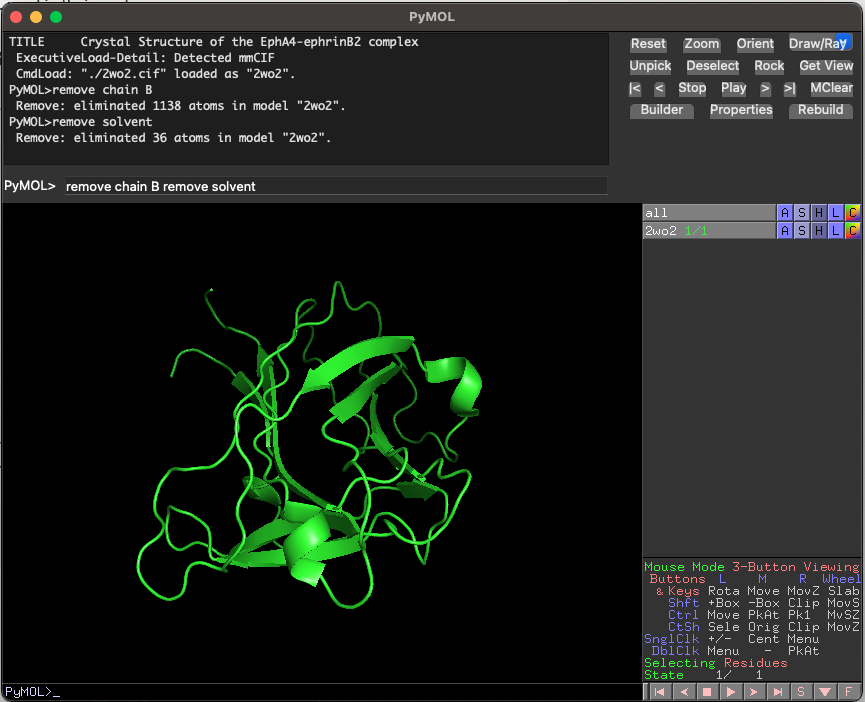
Defining Binding Site Area of EphA4
Open the NRGSuite-Qt plugin from the PyMOL plugin menu and click on the button ‘GetCleft’ (see GetCleft). Click ‘Refresh’, select ‘2wo2’, and press ‘Play’.
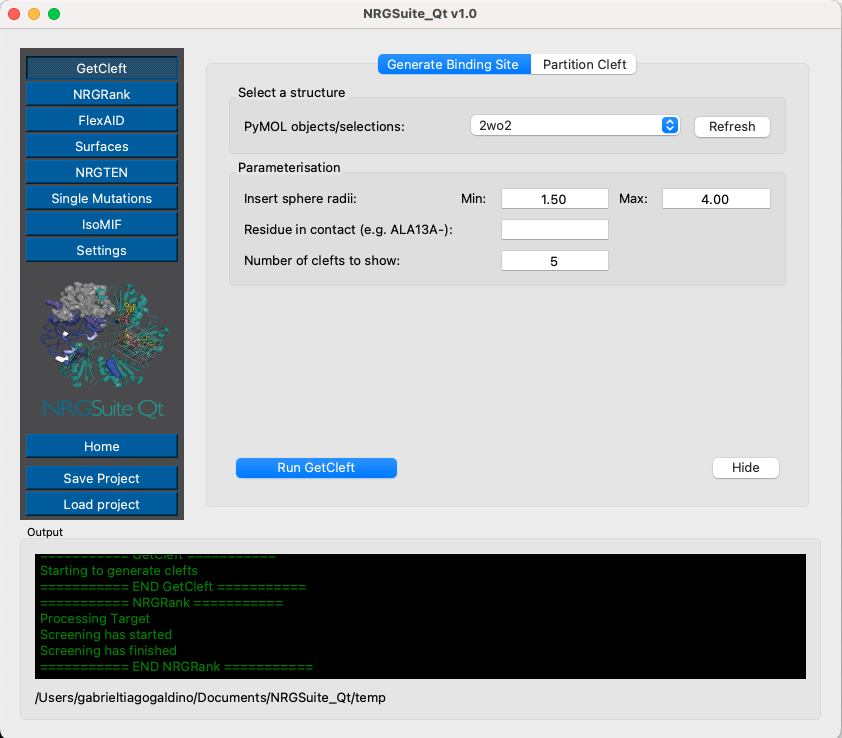
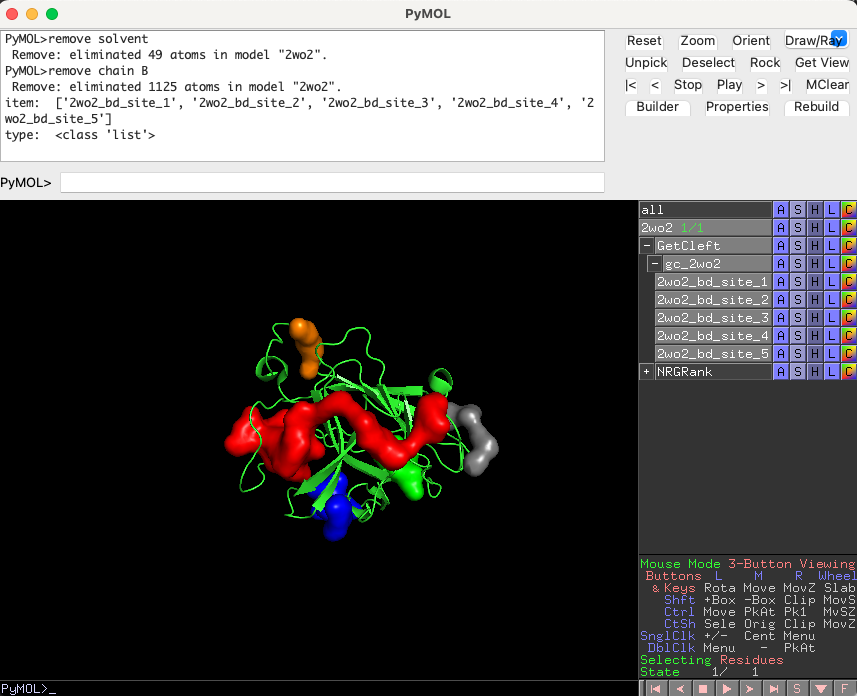
Five clefts should load in the PyMOL interface. The cleft with the largest volume, ‘2wo2_bd_site_1’, is the one we will use for our docking experiments.
Running the FDA-Approved-Drugs Ligand Set on EphA4
In the plugin interface, click the ‘NRGRank’ button in the left corner. Go to the ‘Settings’ tab.
Set the number of ‘Save poses for top n results’ to 20 (so we can have the structures of the top 20 molecules). We expect that nilotinib (code: DB04868), the inhibitor identified in the paper, will be ranked among the best-scored ligands of the ‘DrugBank FDA’ ligand set, so it should appear among the top 20 ligands.
The ligand rotations will remain at 9 (default). The user can choose how much of CPU usage to allow, we recommend to use the default (75%).

Click the ‘Run’ tab. Press ‘Refresh’ on the target list and select ‘2wo2’. Press ‘Refresh’ in the ‘Binding site’ list and select ‘2wo2_bd_site_1’. Press ‘Run’ and wait until the progress bar is full.

The top 20 ligands will be plotted in the PyMOL interface in a group called ‘NRGRank’ also a list of all results will be shown in the tab Results in “NRGRank” menu. Search for ‘DB04868’ in the list and click on it. —this is the pose generated by NRGRank.

You can also access the complete list of results of NRGRank by opening the ‘.csv’ file in the ‘temp’ directory of ‘NRGsuite-QT’ in documents.
To obtain a more realistic pose of the nilotinib/EphA4 complex, we will re-do the docking simulation for nilotinib using FlexAID.
Performing Docking of Nilotinib Using FlexAID
Click the ‘FlexAID’ button in the left corner menu. Go to the ‘Settings’ tab. Set the ‘Number of chromosomes’ to 1000 and ‘Number of generations’ to 1000. Check the box ‘Ligand pose as reference’ to compare FLEXAID and NRGRank poses. We are interested in the best result for FlexAID, so set “Max results” to 1

Go to the ‘Simulate’ tab. Press ‘Refresh’ in the target list, ligand list, and bind-site list. Select ‘2wo2’ as the target, ‘DB04868’ as the ligand, and ‘receptor_sph_1’ as the target cleft.
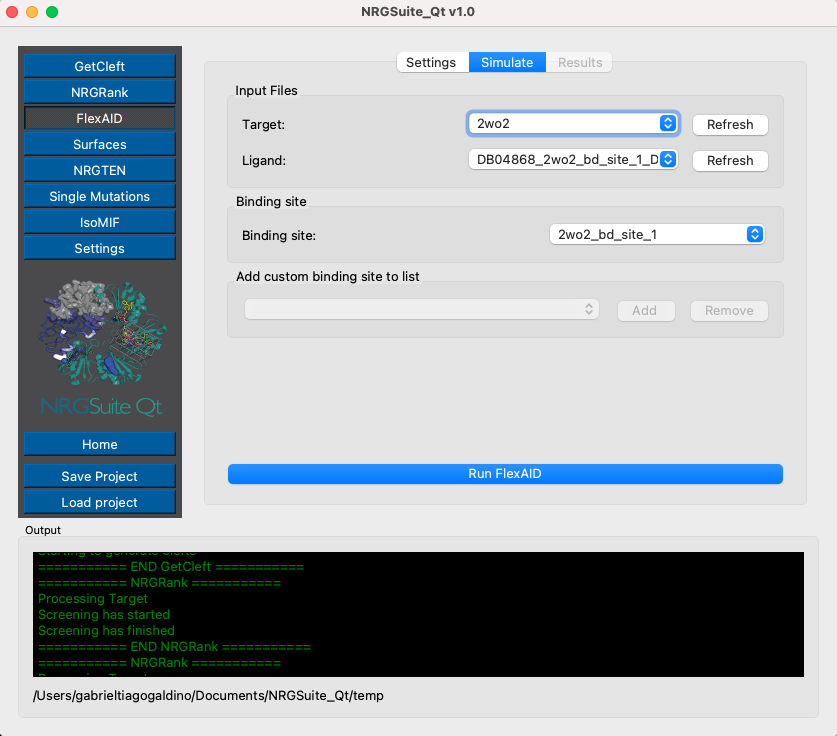
The ‘Results’ tab will open automatically. The progress bar will indicate the progress of the simulation and list the top 1 best ligand pose and it score (CF, most negative -> better scoring) and RMSD in relation to ‘NRGRank’ result.

The top 1 result will be plotted in the PyMOL interface in a group called “FlexAID”
Visualizing Nilotinib/EphA4 Interactions with Surfaces
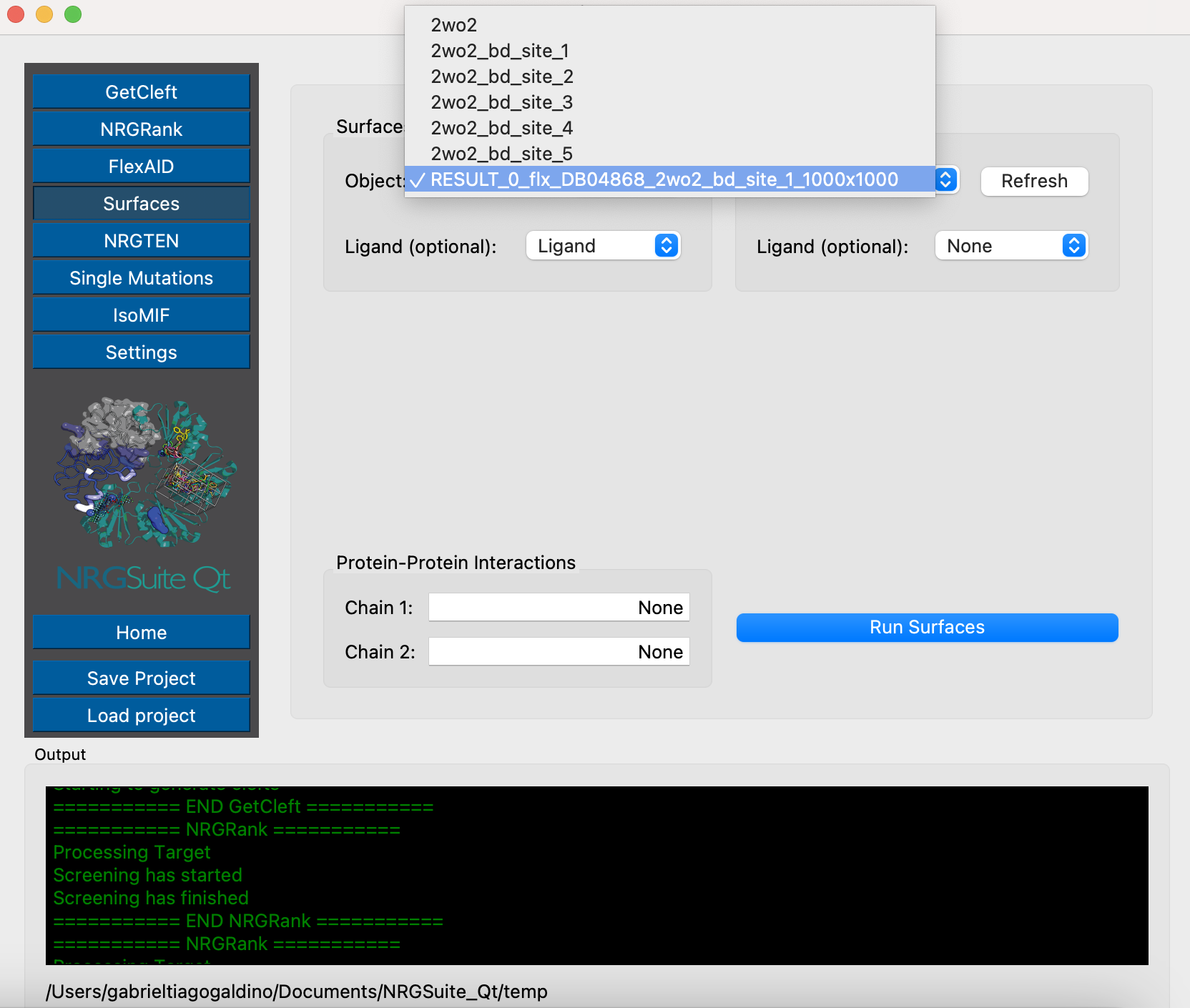
We will now run the Surfaces for the top pose of FlexAID. Make a selection for the ligand:
select Ligand, resn LIG and RESULT_0_flx_DB04868_2wo2_bd_site_1_1000x1000
Click the ‘Surfaces’ button in the menu. Click in ‘Refresh’ on the ‘Surfaces selection:’ area and select ‘RESULT_0_flx_DB04868_2wo2_bd_site_1_1000x1000’. Press ‘Run Surfaces’. Select ‘Ligand’ in the ‘Ligand:’ list and press ‘Run’
A table with all surfaces results can be seen in the tab ‘Results’, press ‘Refresh’ to list all individual results. Select: ‘List_RESULT_0_csv_output’.

A list view of all interactions is shown in the PyMOL interface in a group named: “surfaces_results”. The table in ‘Results’ table is interactive. Individual residues can be selected by clicking the result name. A selection named:’sele_surefaces’ is created with that specific residue.

In the article the authors say: ‘Nilotinib is predicted to form hydrogen bonds with Q71 in the D-E loop and T104 as well as hydrophobic interactions with F154, V157, I163, L166, A193, and V195.’ Also, we can create a selection with the Top N residues by interection absolute value. Type 10 in the ‘TOP N residues:’ field and press the button ‘Interface’. A selection called ‘all_residues’ is created.
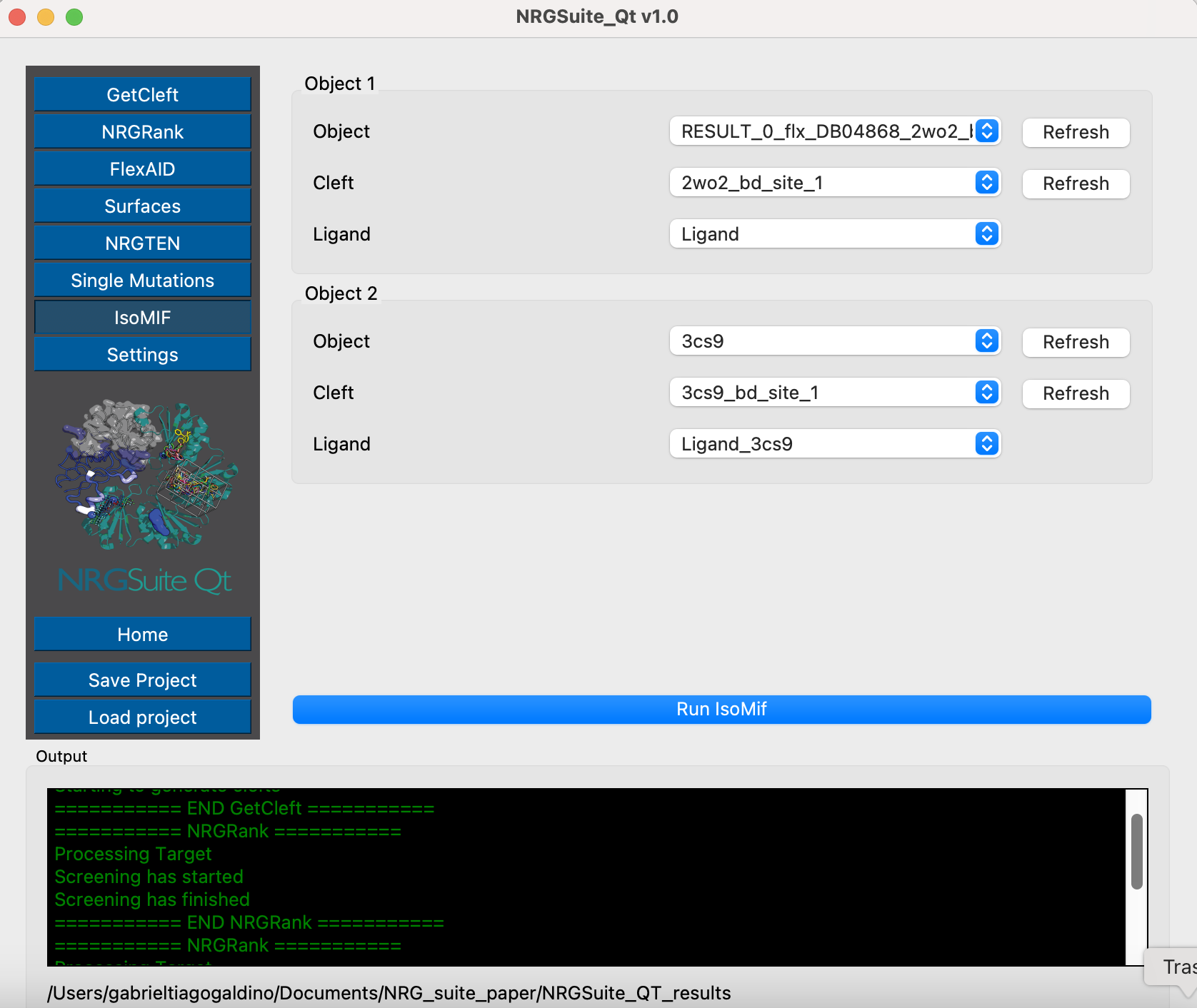
Binding-site comparison between EphA4 and ABL kinase using ISOMIF
A structure of ABL kinase in complex with Nalotinib is available in PDB (https://www.rcsb.org/structure/3cs9). We can use ISOMIF to compare both binding sites of EphA4 and ABL using molecular interaction field to identify geographically and chemically equivalent areas of their binding sites. This can give us an idea of how Nalotinib is capable of biding both proteins and what are the chemical properties important for this process.
To download and make a selection with the ligand in 3cs9, run these commands in PyMOL:
fetch 3cs9, type=pdb1 remove solvent select Ligand_3cs9, resn NIL and 3cs9
Open ‘GetCleft’ menu. Select 3cs9 in the ‘PyMOL objects/selections:’ list and press ‘start’.

The first cleft will be the one containing the ligand in 3cs9. We can now open ISOMIF menu and in the “Load object 1” area select “3cs9” in “Object 1” list, “3cs9_sph_1” in the “Cleft” list and “Ligand_3cs9” in “LIG” list. In the “Load object 2” area select RESULT_0 in the “Object 2” list, “RESULT_0_sph_1” in the “Cleft” list and “Ligand” list. Press “Run”.