Spike Variant (Protein-Protein analysis)
In this tutorial, we will reproduce a few results regarding the evolution of the SARS-CoV-2 Spike protein. We will employ tools available at NRGSuite-Qt to study its conformatioal dynamics, binding affiity to ACE2 and antibody recognition.
Evaluating dynamical effects of mutations
We will firstly evaluate conformational dynamics. For that purpose, we will use 2 Spike structures, one in the 3-down conformation and one in the 1-up conformation.
Open PyMOL and run the command:
fetch 6vxx fetch 6vyb remove hetatm
The D614G substituion became common during the summer of 2020 and was the first widely selected Spike substitution during the COVID-19 pandemic. Let’s evaluate its effects on the Spike dynamics.
First, we need to perform the mutations. For that, we first need to select the residue we want to mutate:
sele resi 614 and 6vxx
Now that the residue in the closed conformation structure is selected, we can mutate it to the desired aminoacid:
We need to do the same for the open state conformation. We first select the residue:
sele resi 614 and 6vyb
Now we can also perform the mutation using the Single Mutations functionality:
Once the mutations are done, we can evaluate their effects on dynamics using NRGTEN. For that, we will run the Dynamical Signature function, first for the closed conformation structure:
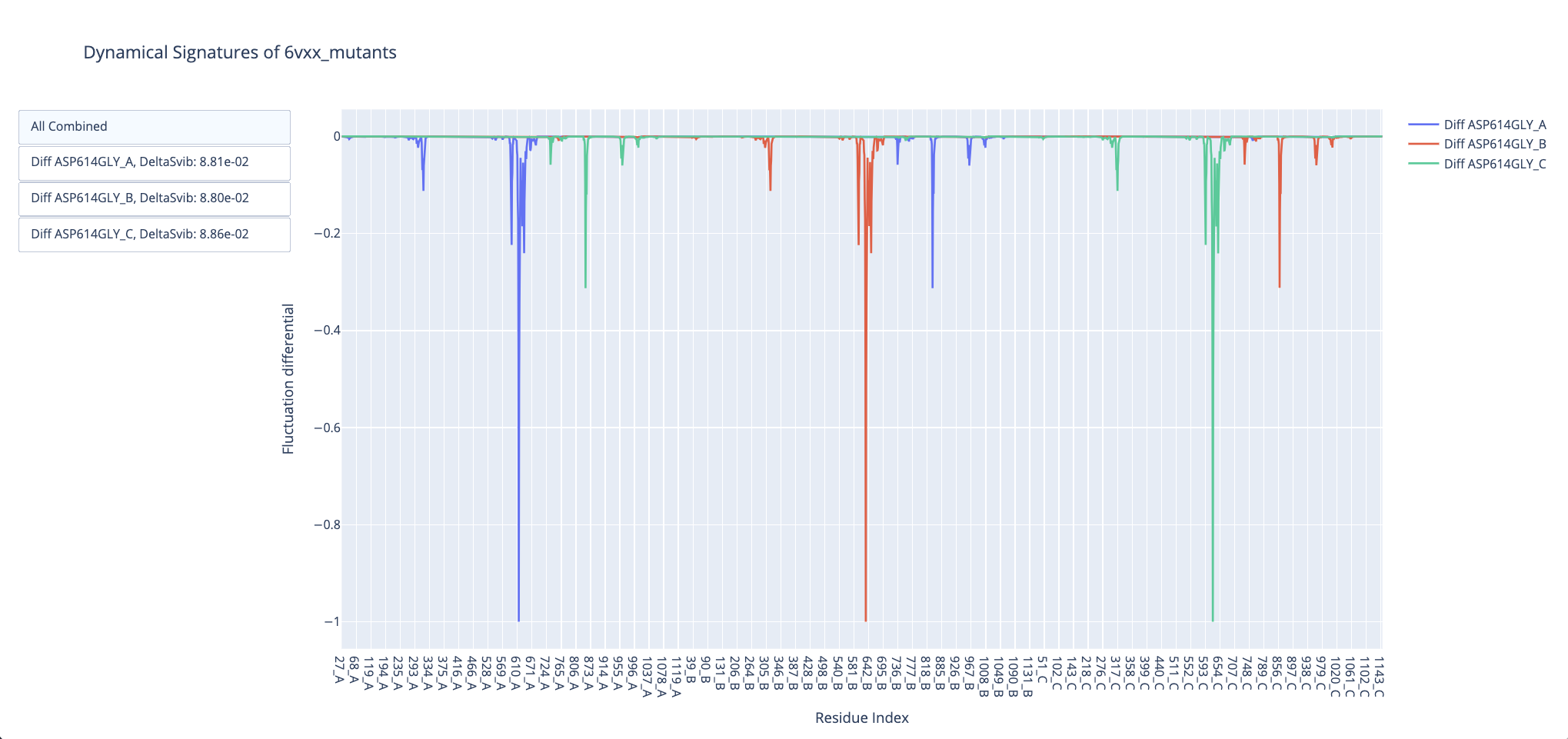
And then for the open conformation structure, so we can evaluate the dynamical effects for both states:

We can see the pattern of increased flexibilty of the closed state and decreased flexibility of the open conformation, particularly around the open Receptor-Binding Domain, which is also observed in the DeltaSvib results, representing the flexibility of the whole structure. These results reproduce the observations from Teruel et al. 2021 for the D614G mutation favoring the open state occupancy, also confirmed by more costly computational methods and experimental observations (Mansbach et al. 2021, Gobeil et al. 2021).
Teruel et al. proceeds to look for the same pattern of effect on flexibility for over 17,000 mutants. Here, we will reproduce two of the main results, for positions 417 and 501.
First, we need to create the mutations. For that, we first select the residue we would like to mutate - residue 417, in this case:
sele resi 417 and 6vxx
We can then run the Single Mutations function:
The same is necessary for the other conformational state:
sele resi 417 and 6vyb
Now that the mutations are done for both conformational states, we can evaluate their effects on dynamics. We first run NRGTEN to get the Dynamical Signature for the closed conformation:
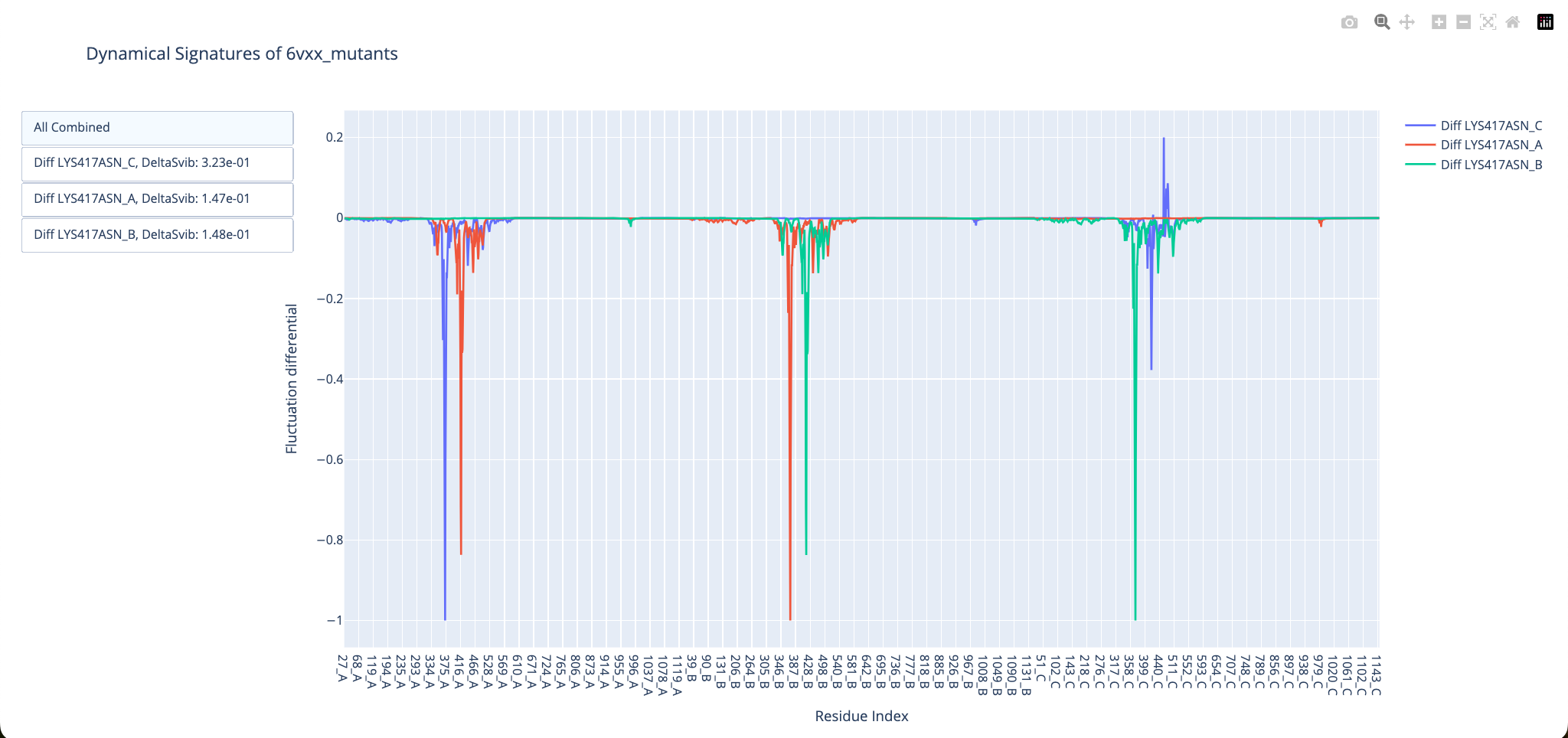
And do the same evaluation for the open conformation:

We see that K417N has similar effects to D614G, making the closed conformation more flexible around the Receptor-Binding Domain, and the open Receptor-Binding Domain becomes more rigid. We can also see this in the DeltaSvib results, in which the evaluation for the closed conformation gave us positive results, meaning a more flexible mutant, while the open conformation gave us negative results, meaning less flexible mutants.
We can make the same evaluation for mutation N501Y, starting by selecting the residue:
sele resi 501 and 6vxx
We can then run the implementation of Modeller to perform the single mutation:
And now we repeat the same process for the open conformation structure, selecting the residue and performing the mutation:
sele resi 501 and 6vyb
Now that the mutations are done for both conformational states, we can evaluate their effects on dynamics using NRGTEN. First, let’s run the Dynamical Signature for the closed conformation:
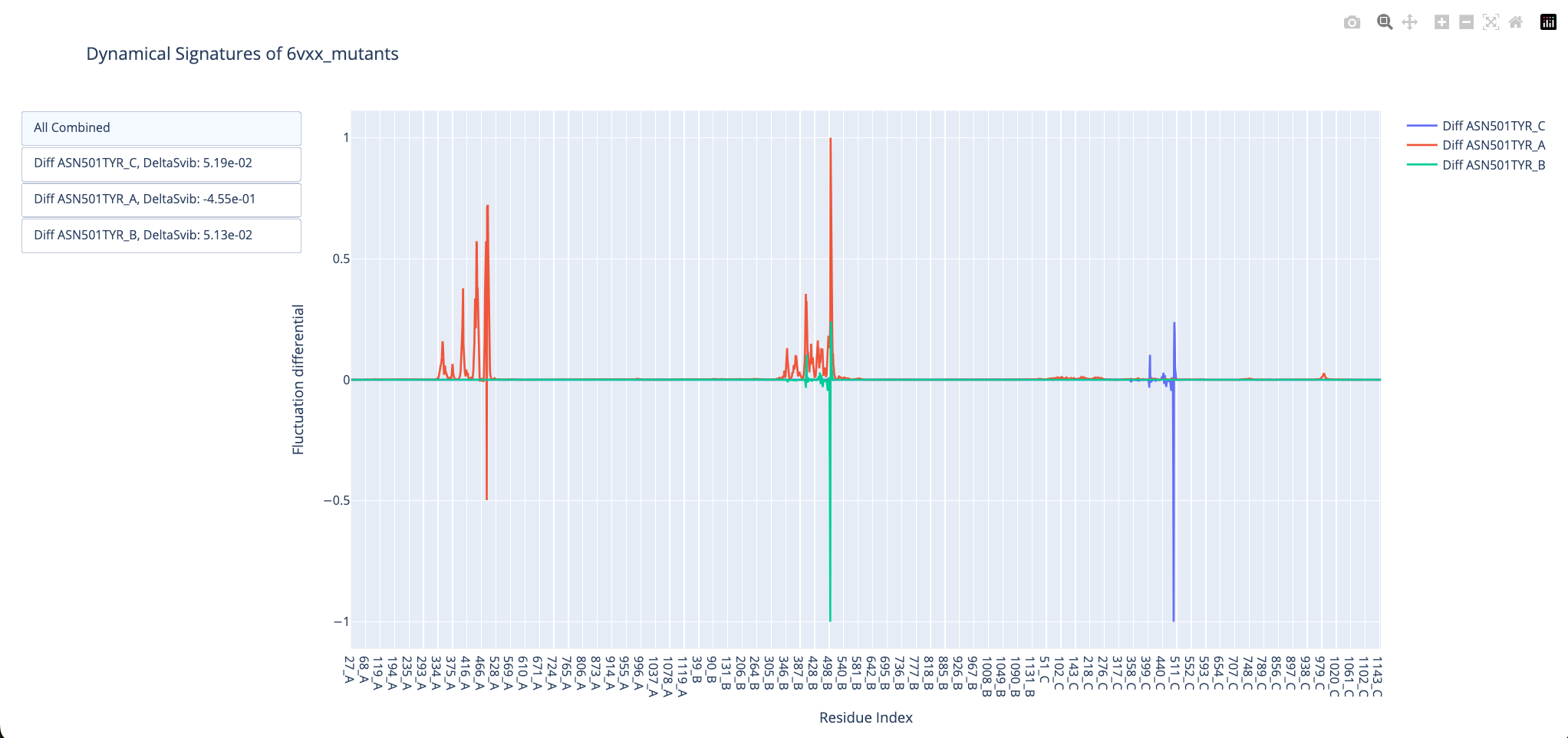
And we can do the same thing for the open structure:
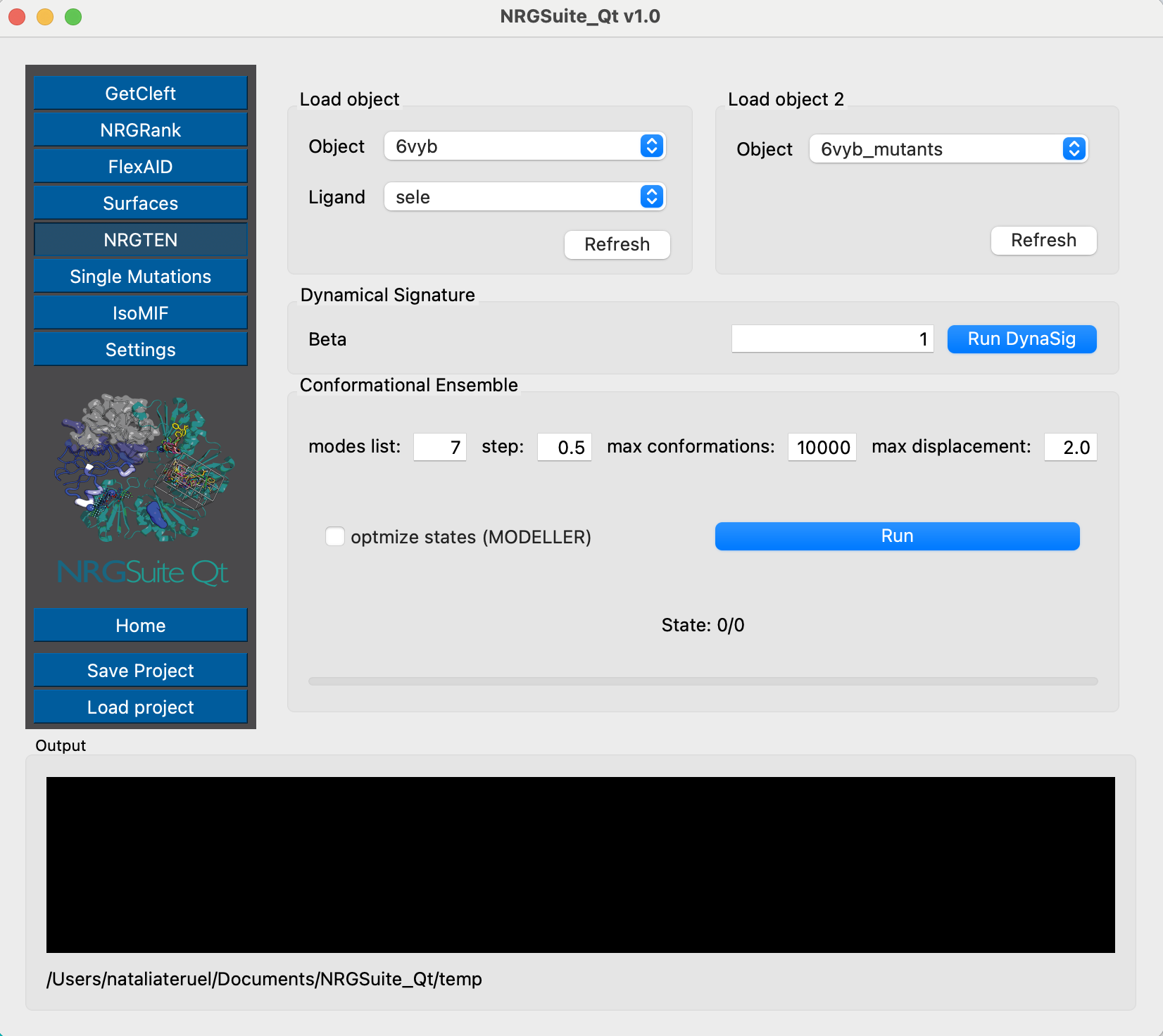

The mutation N501Y also makes the closed conformation more flexible and the open Receptor-Binding Domain more rigid. This reproduces the predictive results from Teruel et al. 2021. The dynamical effects of these mutations have also been shown experimentally (Gobeil et al. 2021).
Evaluating the effects of mutations on ACE2 interactions
For the evaluation of the interaction with the receptor ACE2, we will need a structure in complex with the receptor:
fetch 6m17 remove 6m17 and (chain A or chain C or chain D or chain F) remove hetatm
The mutation N501Y is known for increasing interaction with the receptor ACE2 (Starr et al. 2020, Sergeeva et al. 2023, Tian et al. 2021, Laffeber et al. 2021, Moulana et al. 2022). Let’s see if we can reproduce these results!
We first model the mutation in the complex structure with ACE2. For that, we need to select residue 501:
sele 6m17 and chain E and resi 501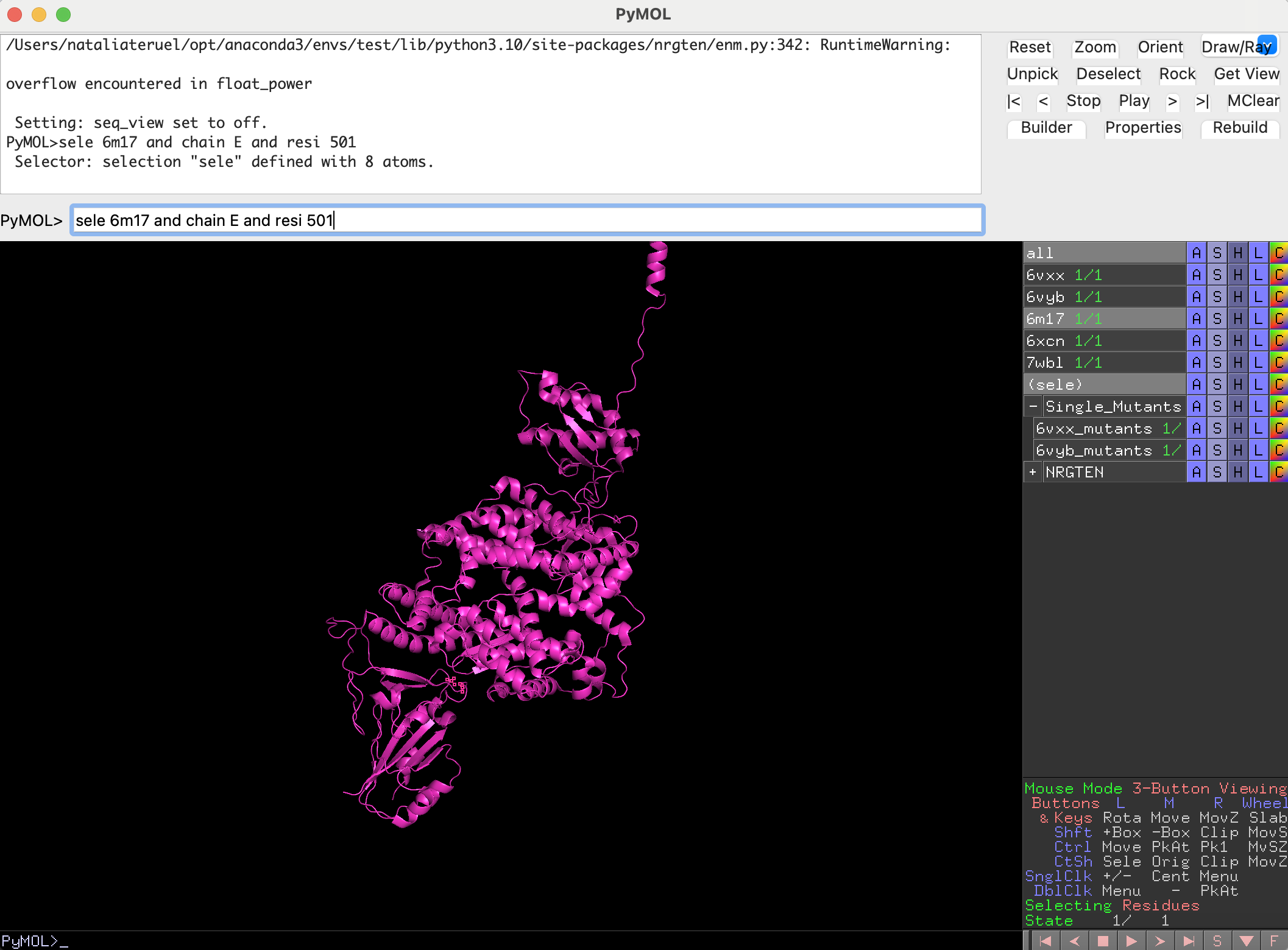
After the selection, we can run Modeller using the Single Mutations function:
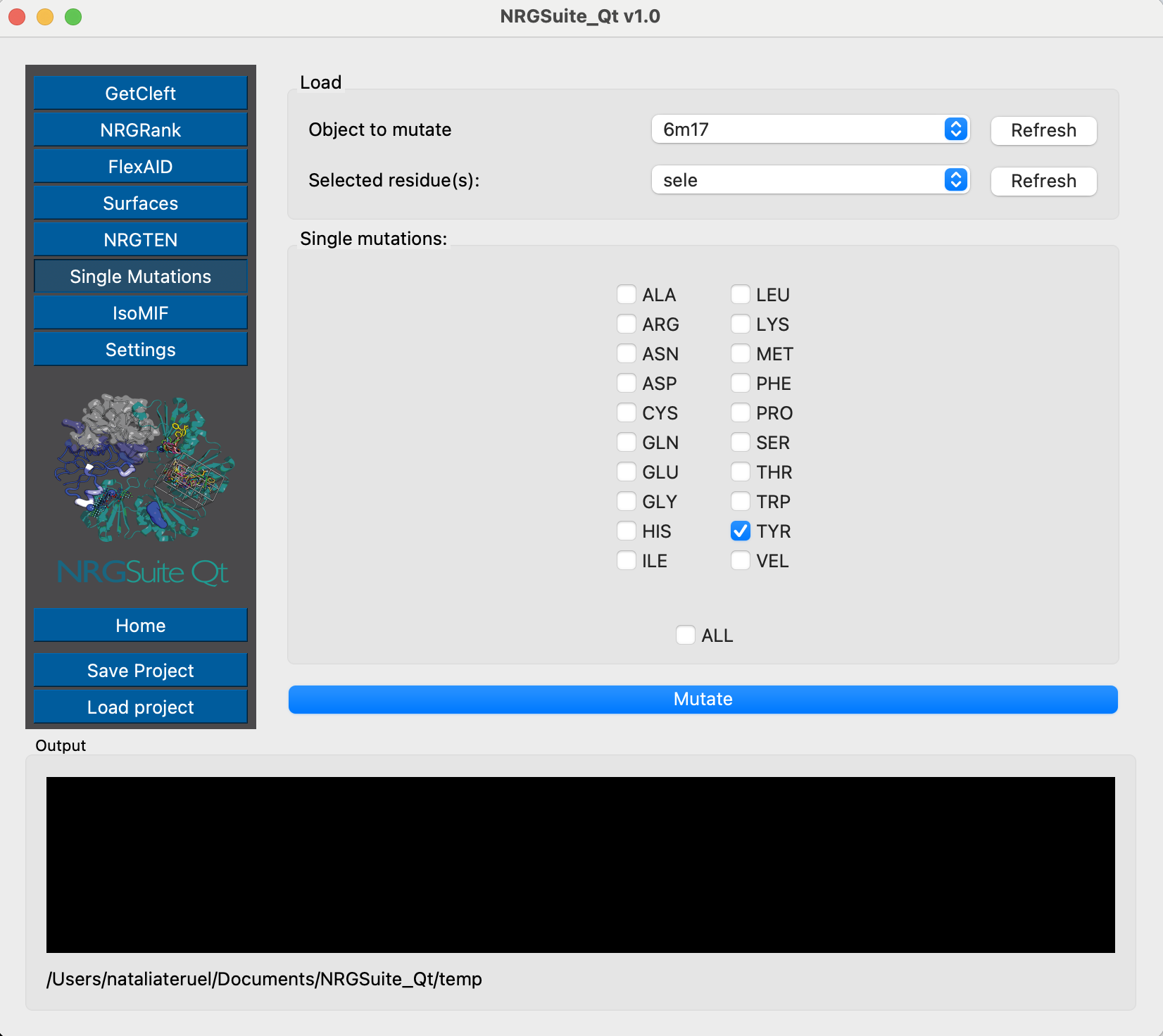

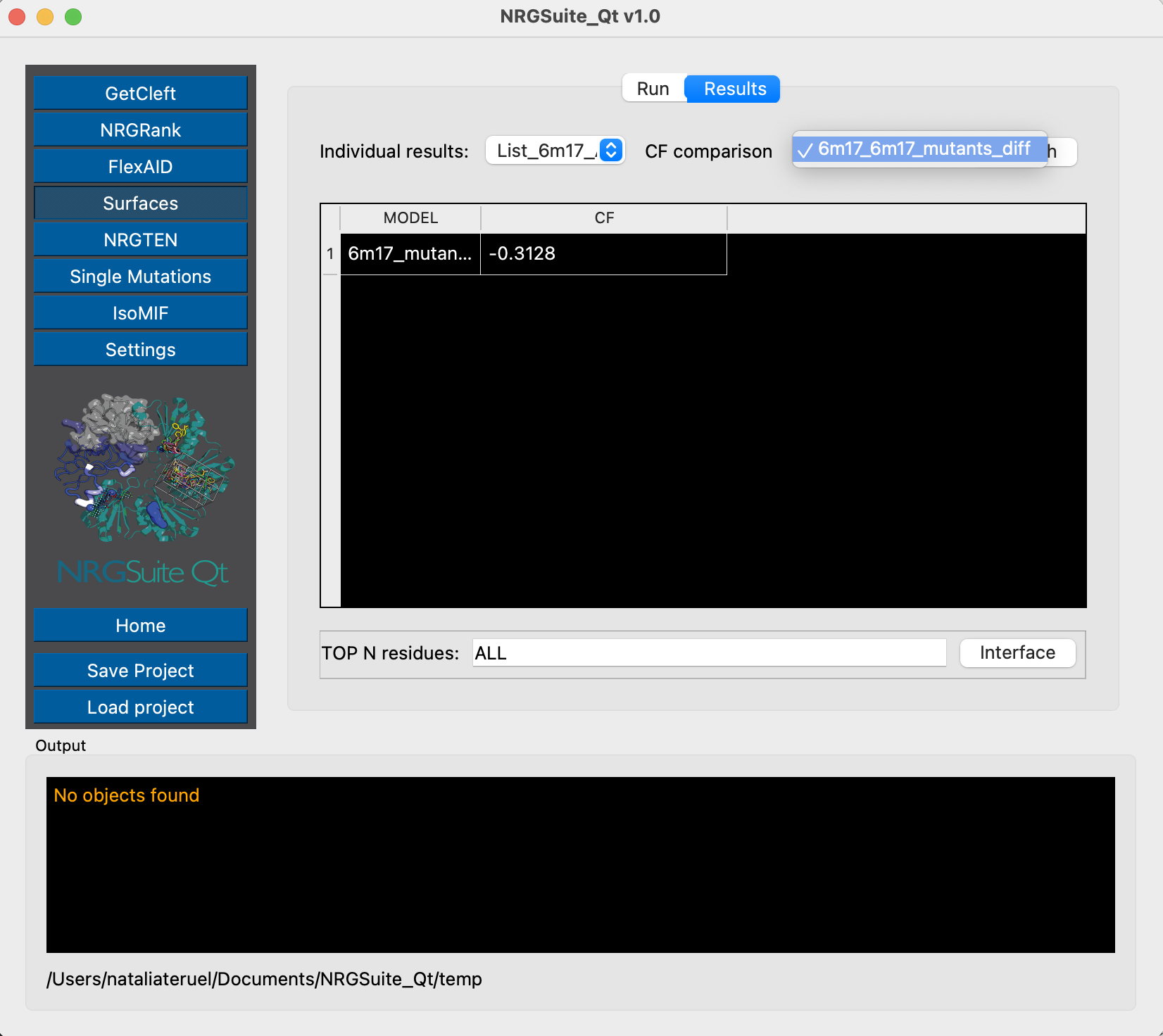
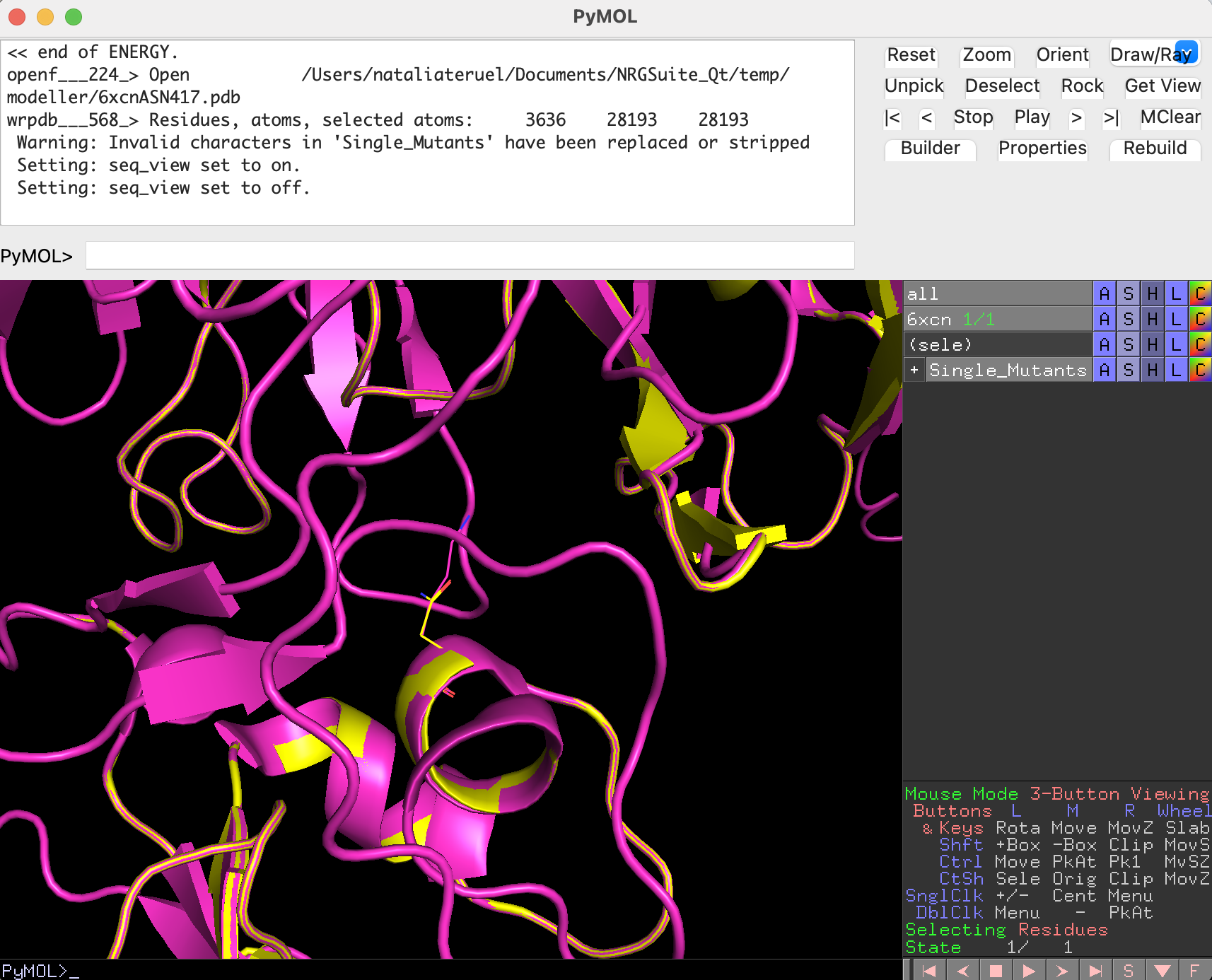
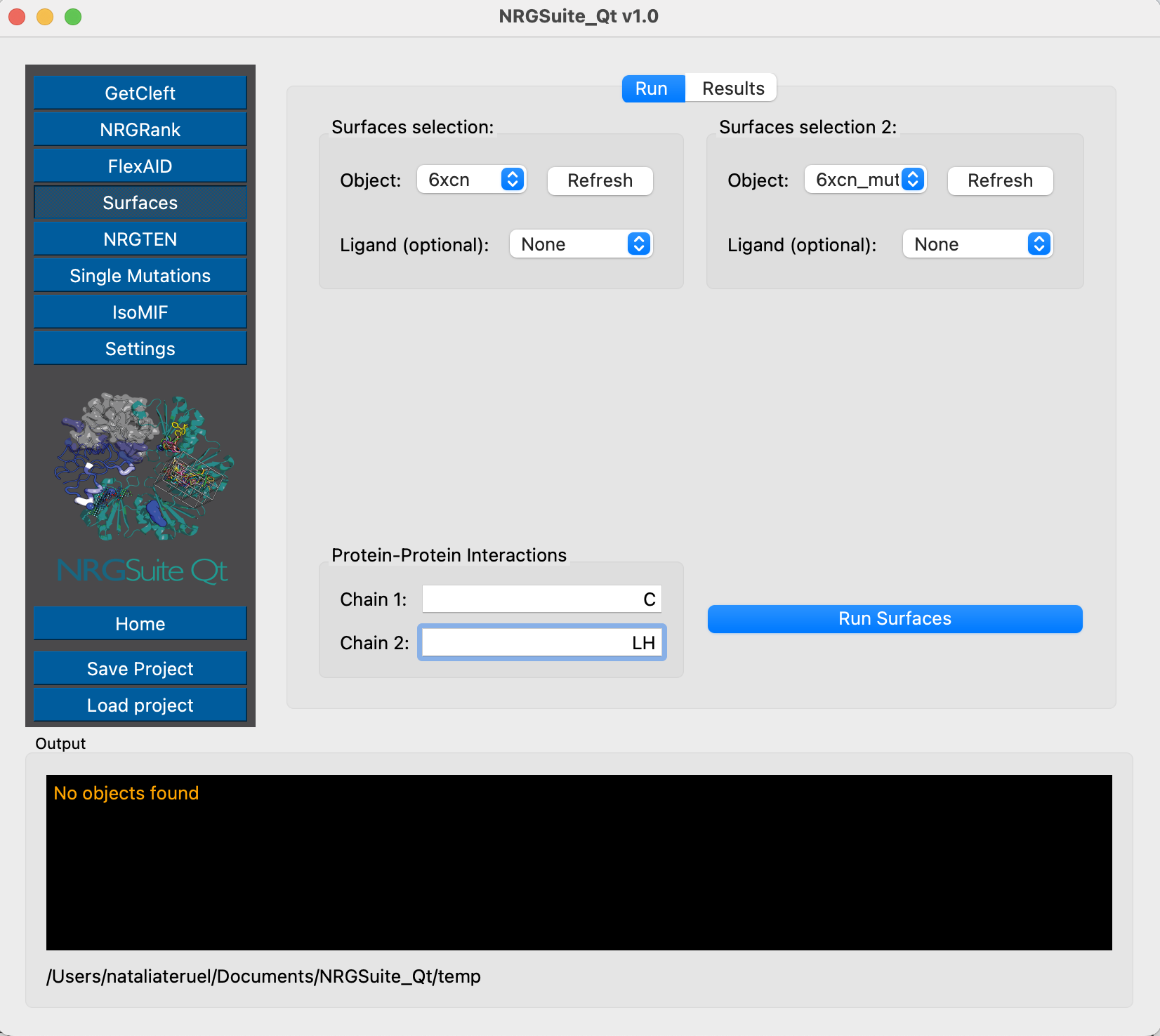
Chain E represents the Receptor-Binding Domain of the Spike protein, and chain C is the ACE2 protein. We can now evaluate their per-residue interactions by using Surfaces:
If we look at the difference in total CF, we see an increase in binding affinity of 0.31 kcal/mol due to the modeled mutation.
Evaluating the effects of mutations on immune recognition
We can also evaluate immune recognition, here represented by interactions with antibody C105. For that, we need a structure of Spike interacting with this antibody:
fetch 6xcn remove hetatm
The K417N substition is in a very important epitope of antibody recognition. Deep mutational scanning (DMS) results show that it affects the recognition of many antibodies, among which the C105 antibody (Greaney et al. 2022, Cao et al. 2023). We can try to reproduce these results evaluating the complex structure.
We start by modeling the mutation. For that, we first select residue 417:
sele 6xcn and chain C and resi 417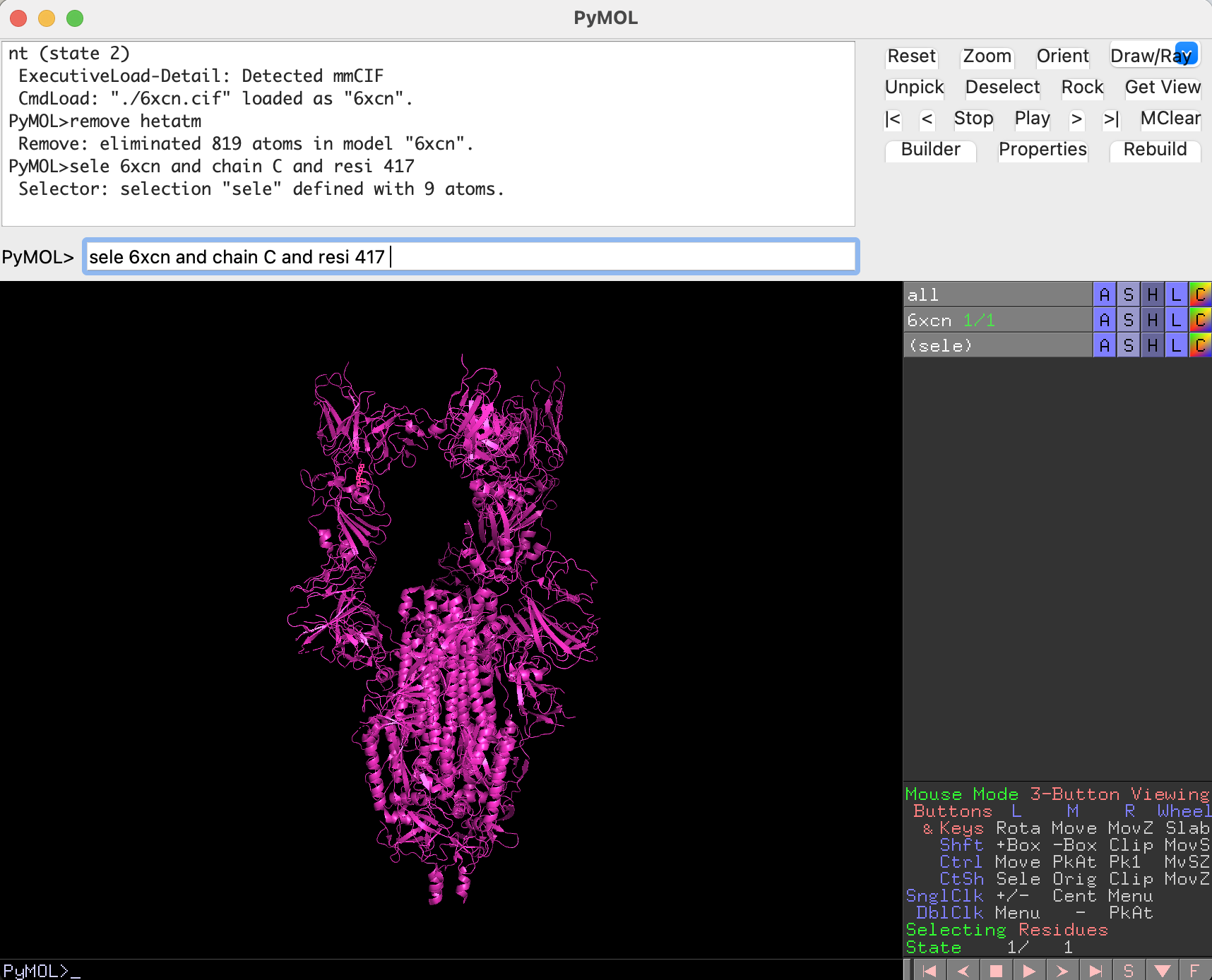
And perform the mutation in the selected position:
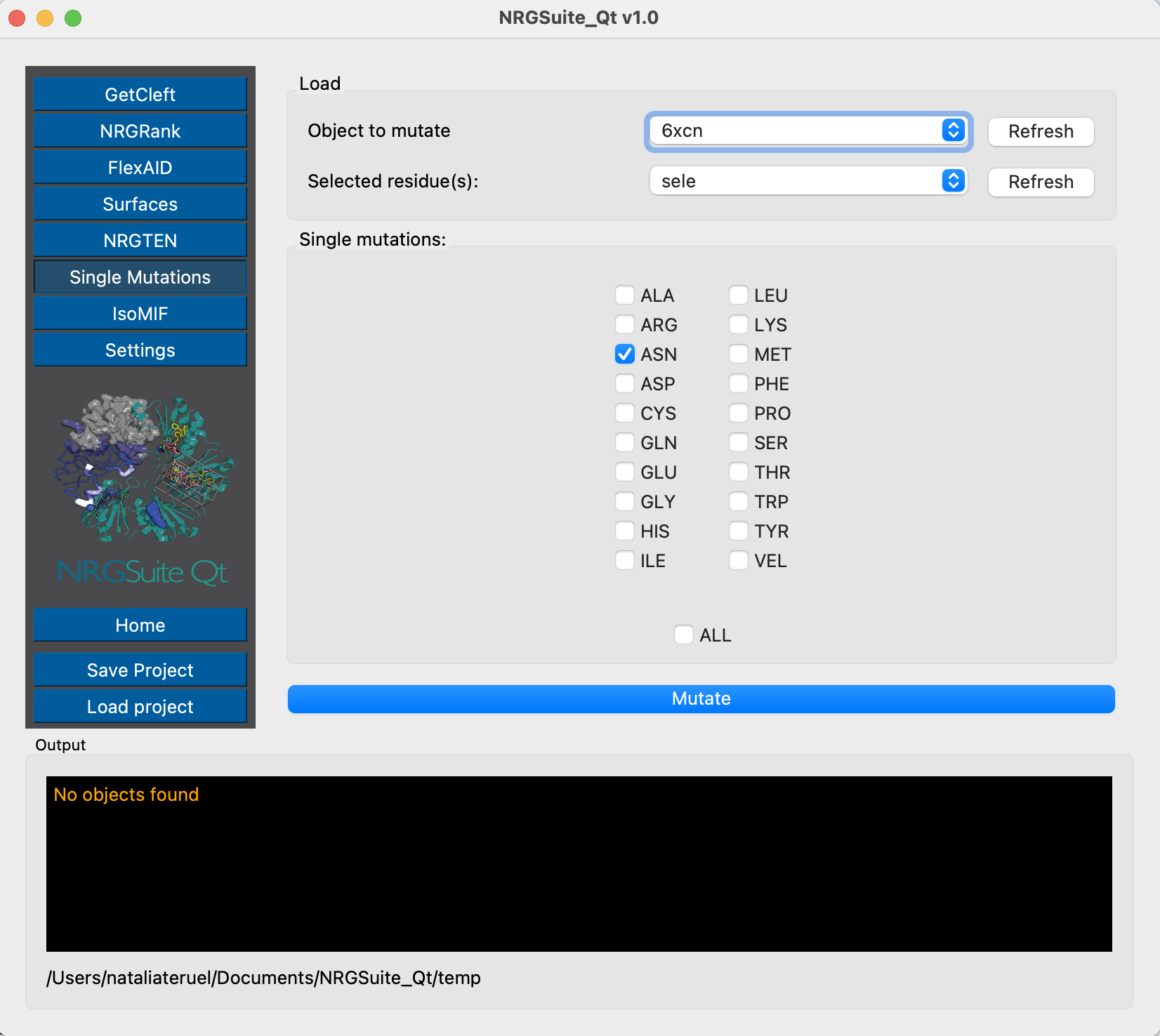
And we can evaluate the interactions of the wild-type and mutant Spike structures with C105 using Surfaces - the chain representing Spike is chain C and the antibody is built by the light chain L and the heavy chain H.
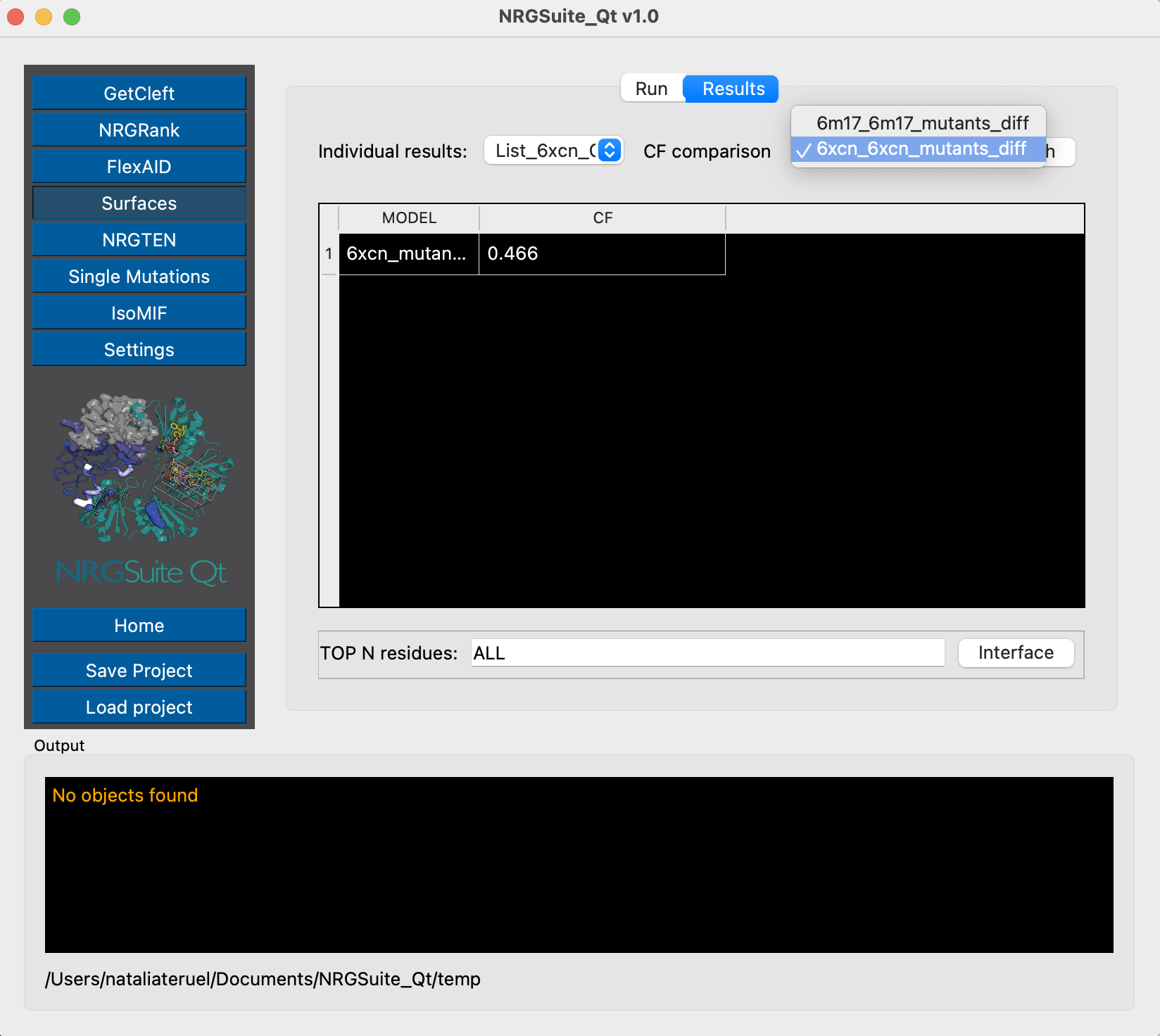
We see that the mutation K417N lowers the binding affinity in 0.47 kcal/mol, in agreement with escape evaluations.
Conformational ensembles
We will use conformational ensembles to evaluate the interactions with ACE2 for the Omicron variant. For that, we will use a PDB structure of the Omicron Receptor-Binding Domain in complex with the receptor ACE2:
fetch 7wbl remove hetatm
Evaluating interactions based on a single or a few structures is inherently limited by the lack of representation of structural variability. Proteins are dynamic entities with flexible backbones and side chains. To more accurately assess complex interactions, it is essential to account for this structural variability.
One way to achieve this is by employing conformational ensembles. In this study, we analyze the interactions between the Omicron Spike protein Receptor-Binding Domain and the human ACE2 receptor. When the Omicron variant was first characterized, numerous publications examined the effects of its mutations on ACE2 binding using various experimentally solved structures. However, due to the inherent structural variability among these structures, the conclusions reached by different studies often diverged (McCallum et al. 2022, Han et al. 2022, Mannar et al. 2022).
To simulate the structural variability, we first need to create a conformational ensemble:
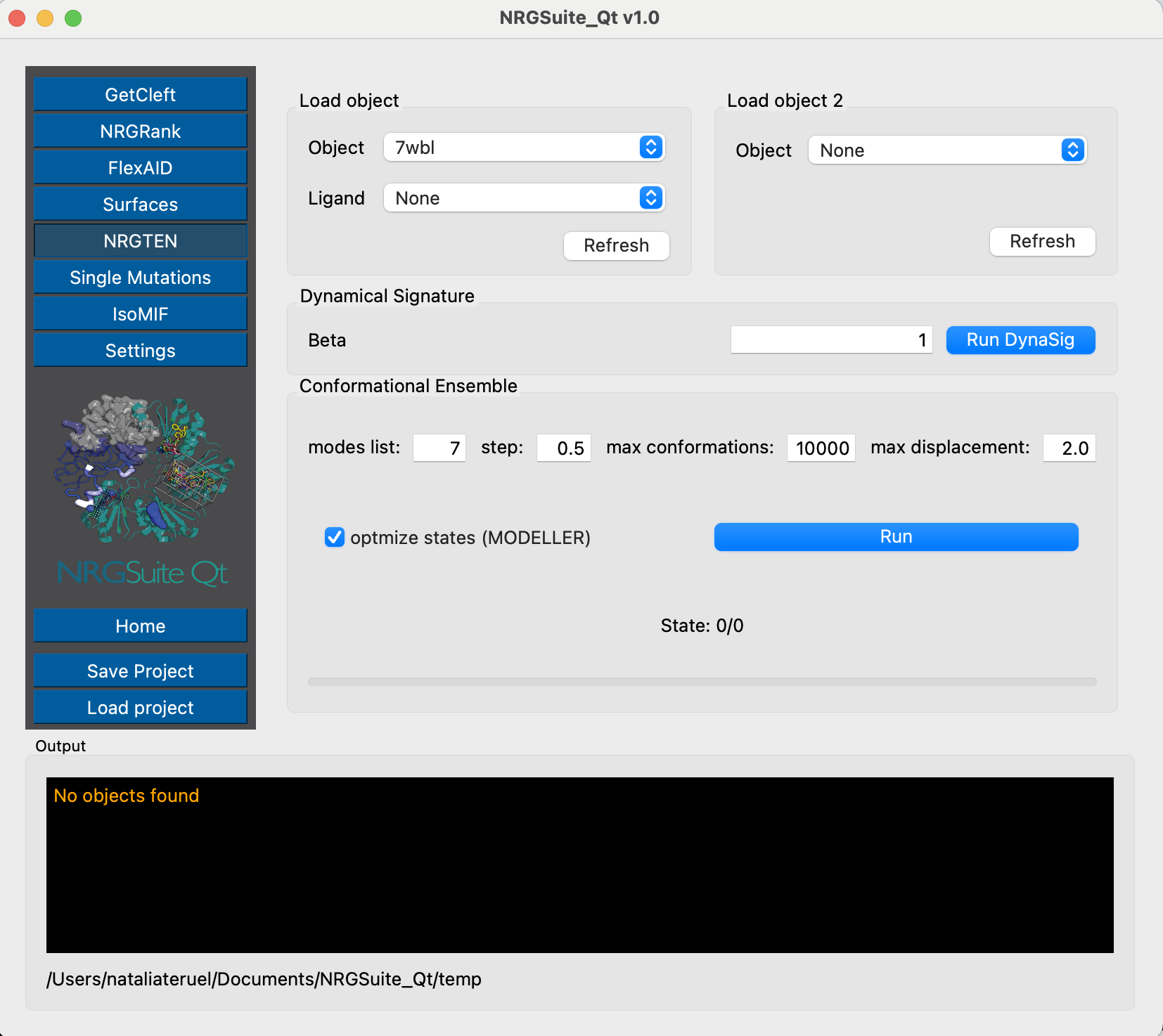
It is important to notice that the modeller optimization changes the numbering of the residues. To properly compare the per-residue interactions, we need to fix the numbering first.
alter (chain A and 7wbl_ensemble), resi=str(int(resi) + 19) alter (chain B and 7wbl_ensemble), resi=str(int(resi) - 264)
Once the multiple states are created and the residue numbering is correct, we can separate them into different objects in order to evaluate them separately:
split_states 7wbl_ensemble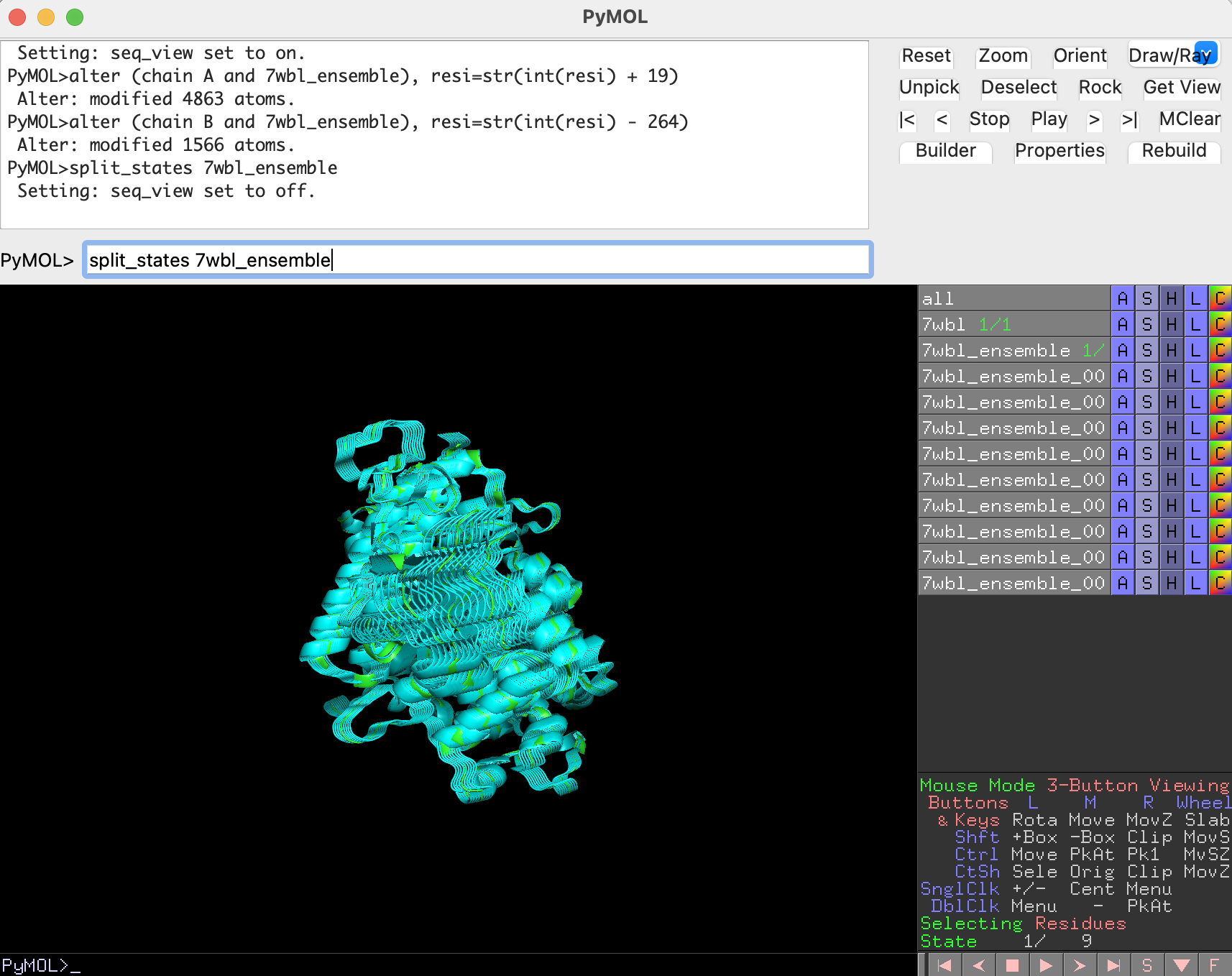
We can then use Surfaces to check the interactions for each one of those objects, and understand the possible interaction variation that comes with the structural variability. The Receptor-Binding Domain is represented by chain B, and ACE2 is represented by chain A.
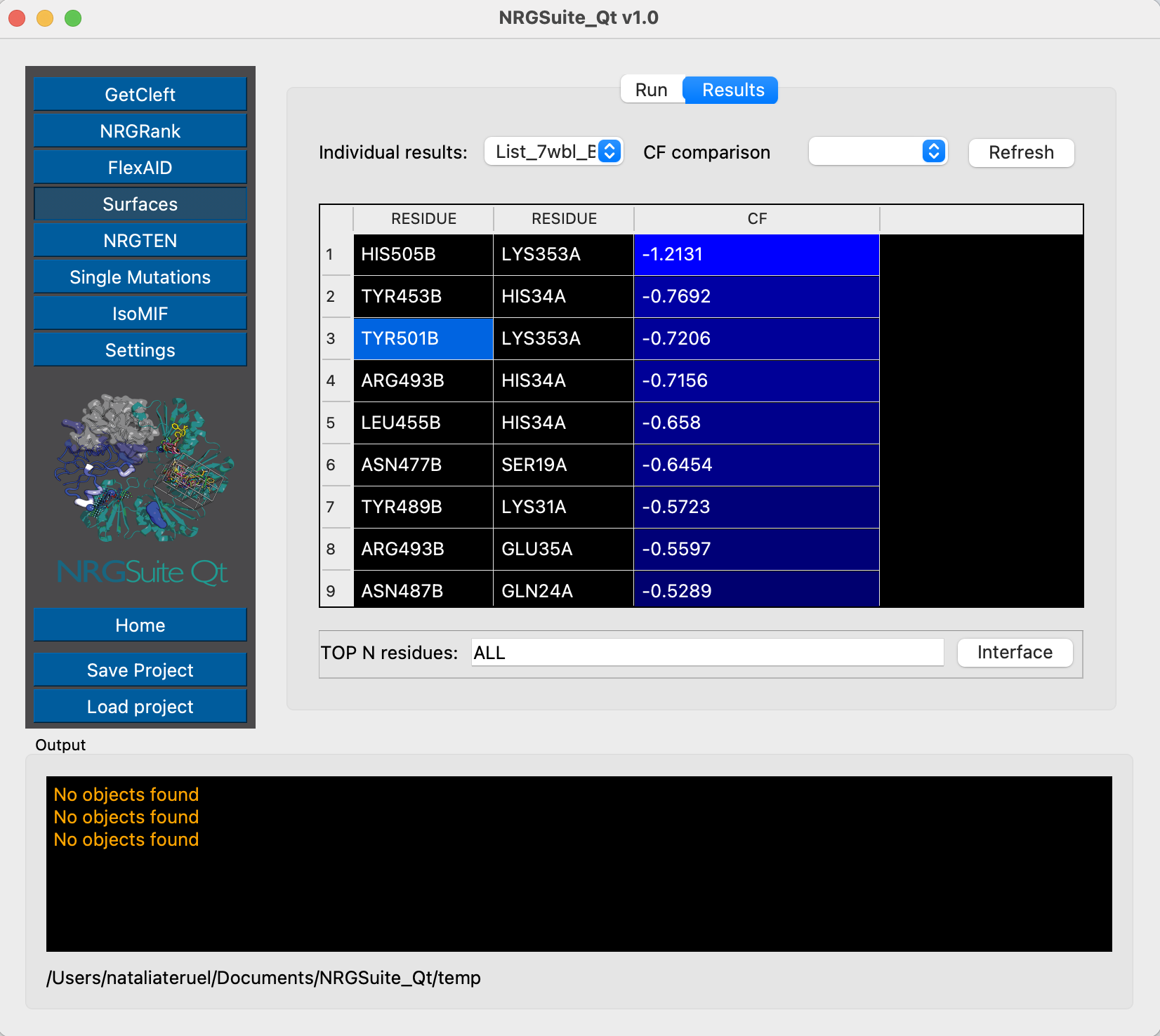
We have already characterized residue Y501 as an important residue for ACE2 interaction. From Surfaces results we can see that in the original Omicron structure the CF of interaction between the residue Y501 from Spike and K353 from ACE2 is of -0.72 kcal/mol. We can evaluate now each of the states of the conformational ensemble to see possible variations in this interaction. First, let’s do it for state 1:
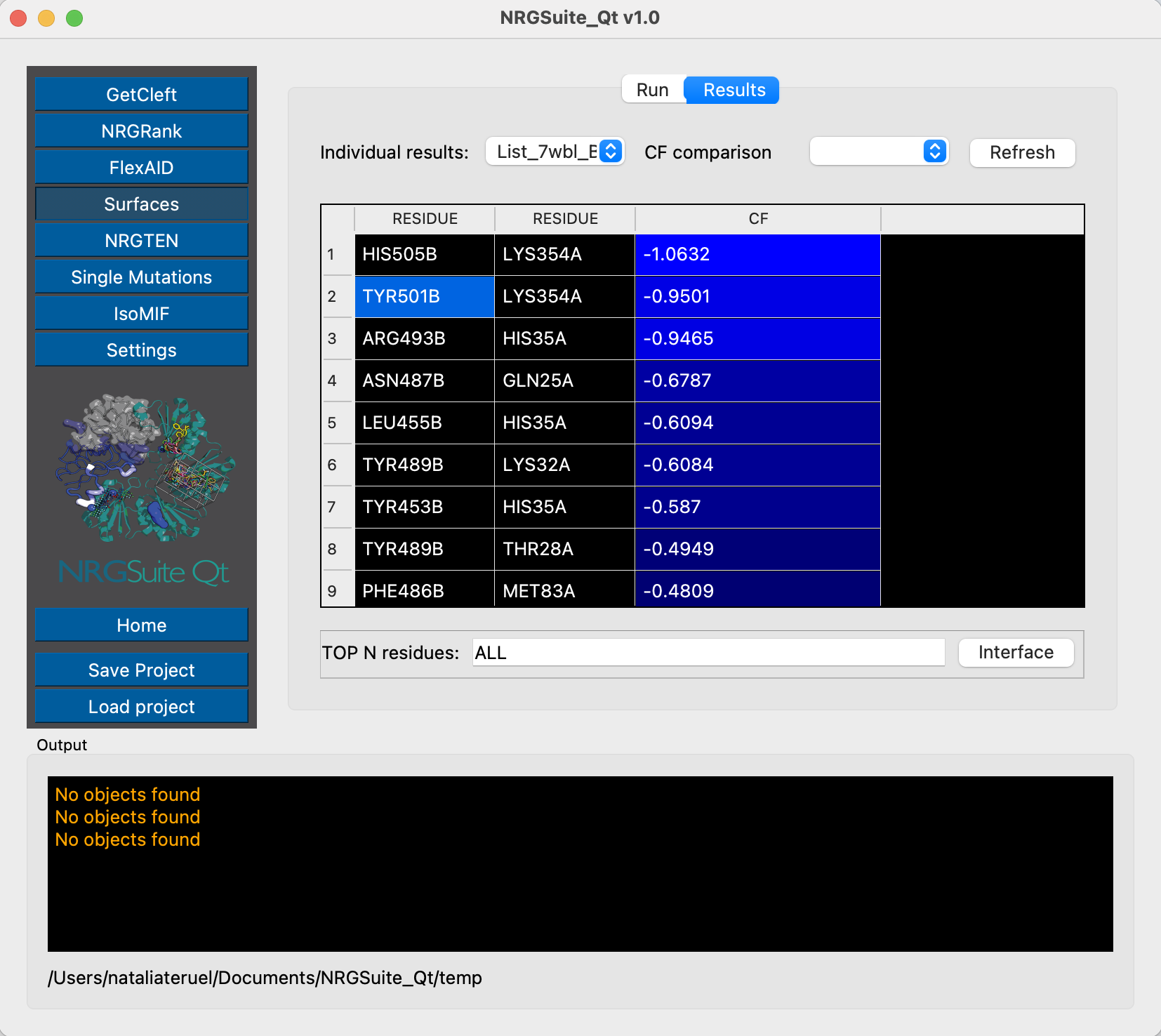
And now for state 9:
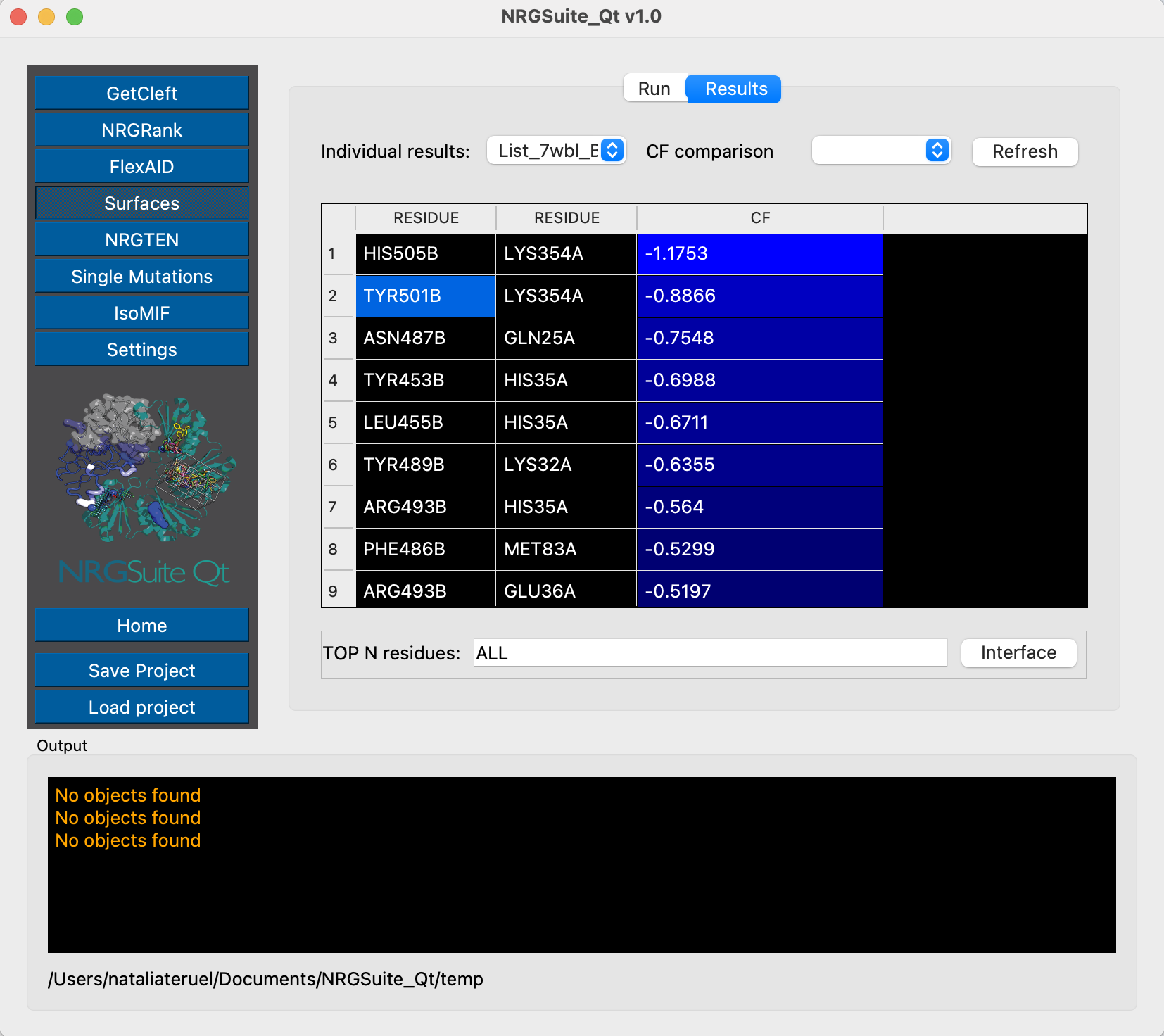
These two examples, of state 1 and state 9, show, respectively, the same pairwise interaction with binding affinities of -0.95 kcal/mol and -0.88 kcal/mol, showing how the conformational variability may impact the results for binding interactions, which can justify the differences from evaluations performed with single structures. The use of conformational ensembles can help overcome this issue.