Surfaces
Surfaces (https://academic.oup.com/bioinformatics/article/39/10/btad608/7288175) comprises a collection of Python scripts that provide rapid means to assess protein interactions by analyzing the atomic surface areas in contact and the types of atoms involved in the interaction. Surfaces is fully implemented in NRGSuite-Qt and can be applied to Protein-Protein and Protein-Ligand interaction calculations.
Settings
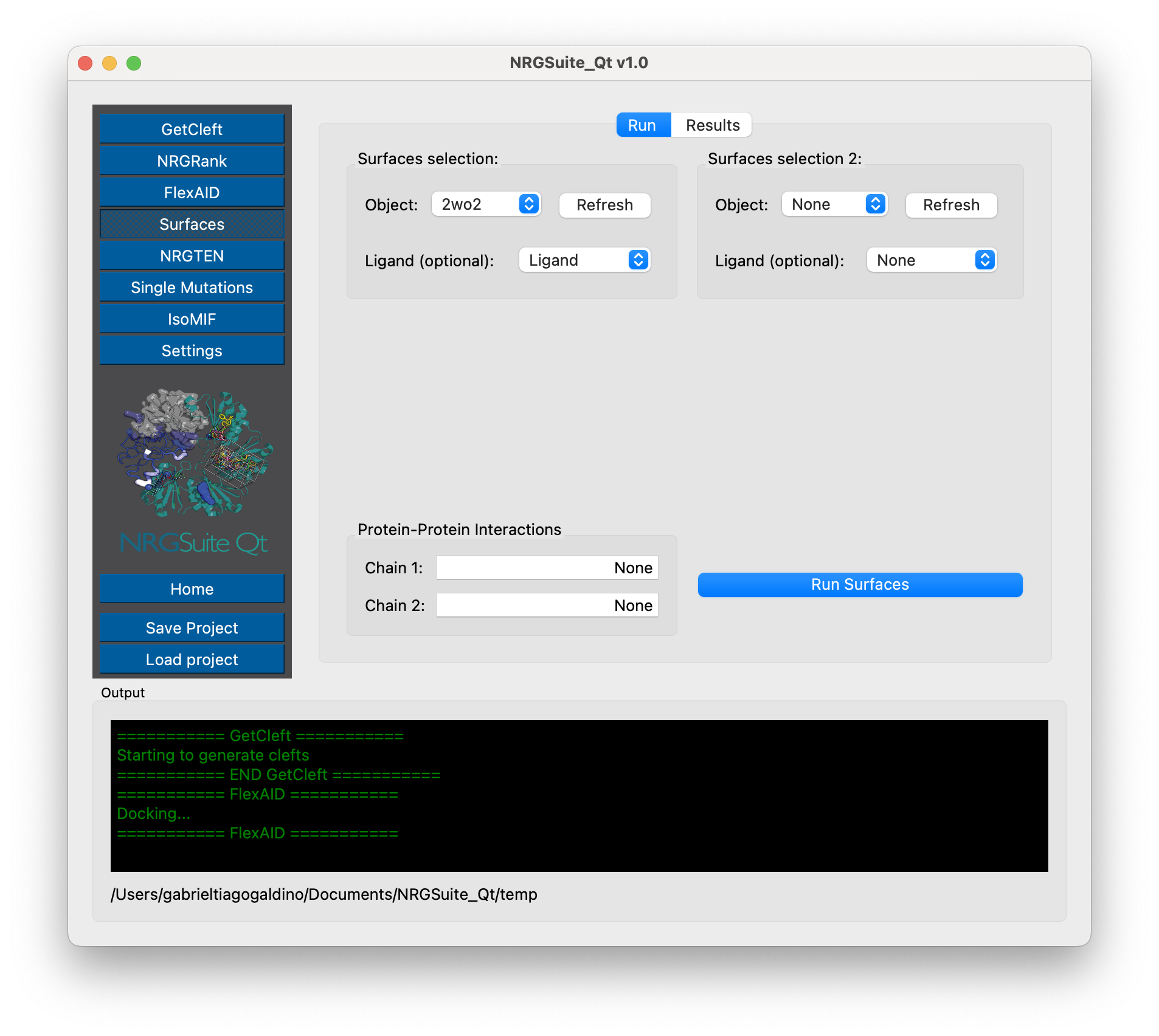
Press ‘Refresh’ in the “Surfaces selection” area.
Select the object containing the protein of interest in the “Object” list.
If running for Protein-Ligand interactions: Select the selection containing the ligand in the “Ligand (optional)” list. All other fields should be marked as “None”. By default, if a ligand is specified, the chain information in the “Protein-Protein Interactions” section is ignored.
Note
In order to run Surfaces to calculate Protein-Ligand interactions, the ligand and the target must be loaded in PyMOL in a single object. A selection containing the ligand must be created.
If running for Protein-Protein interactions: The “Ligand (optional)” list must be marked as “None”. In the “Protein-Protein Interactions” section the user must specify one or multiple chains as “Chain 1” and as “Chain 2”, those chains must be one letter or all letters representing all chains in the group (i. e. “A” for “chain A” or “ABC” for a group of three chains A, B and C).
Note
In order to run Surfaces for Protein-Protein interactions all chains must be loaded in the same object.
If the user specifies “ABC” as “Chain 1” and “DE” as “Chain2”, that will provide a calculation of the interactions between residues of the first group of chains (A, B and C) and residues of the second group of chains (D and E).
For more information refer to the Surfaces manual: https://surfaces-tutorial.readthedocs.io/en/latest/Protein-protein.html
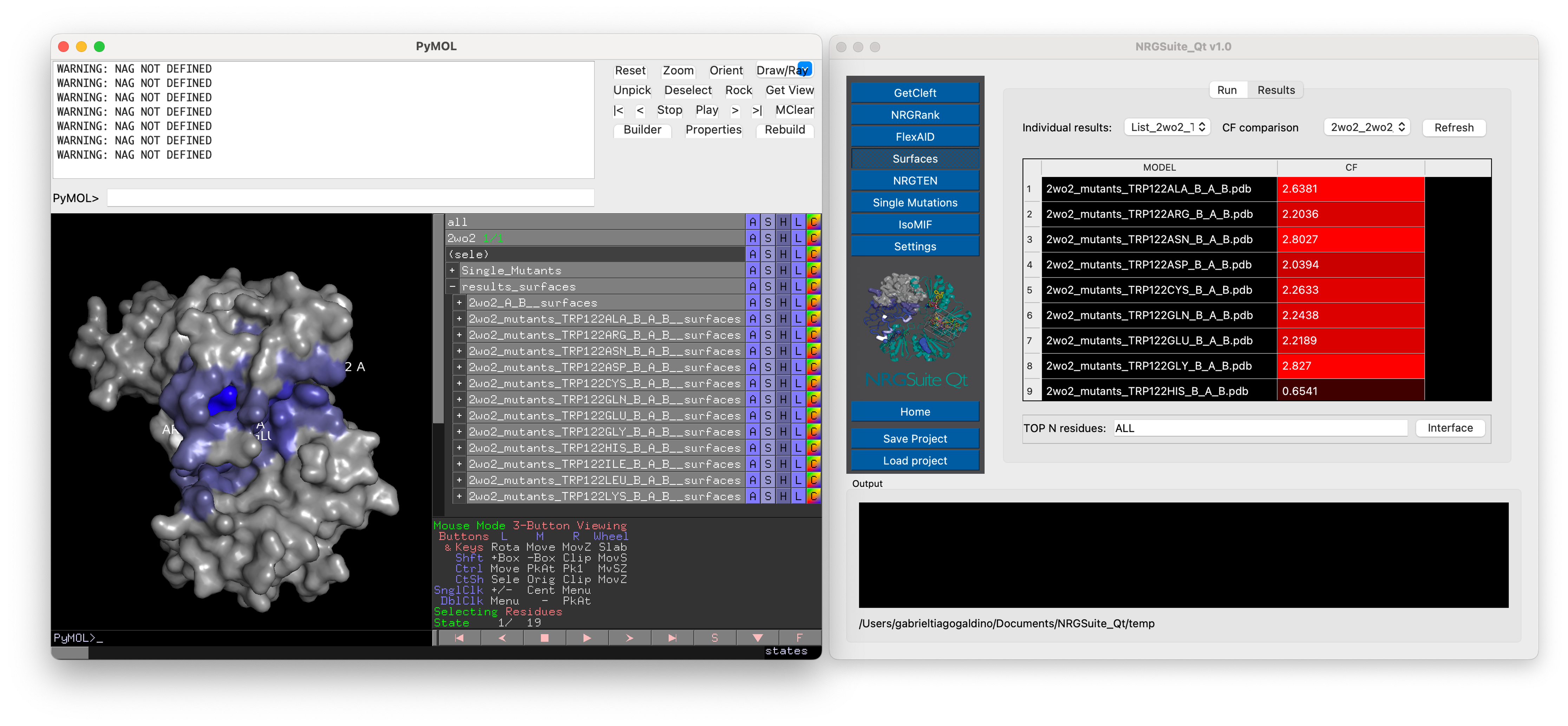
Surfaces also supports the comparison between mutants and wild type. For that all mutants should be present as states of a second object. This object can be generated with the “Single Mutations” functionality (see Single Mutations) or the “Multi-state object Manager” in the “Settings” menu (see Settings).
Select the object containing all mutants in the “Object” list in the “Surfaces selection 2” section.
If analysing the difference of interactions between mutants and a ligand, a selection containing the ligand in the multi-state object should be created. Choose the selection containing the ligand in the multi-state object in the “Ligand (optional)” list in the “Surfaces selection 2” section.
If analysing the difference of Protein-Protein interactions between mutants, the “Ligand (optional)” list should be marked as “None” in both “Surfaces selection” sections. The chains or group of chains to be analysed in all mutants should match the ones in the wild type.
Press “Run Surfaces” to initiate the calculation.
Results
The calculated per-residue interactions, residue(target)/atom(ligand) for Protein-Ligand interactions and residue/residue for Protein-Protein interactions, will be shown in the “Results” tab that will open automatically once the calculation is done.
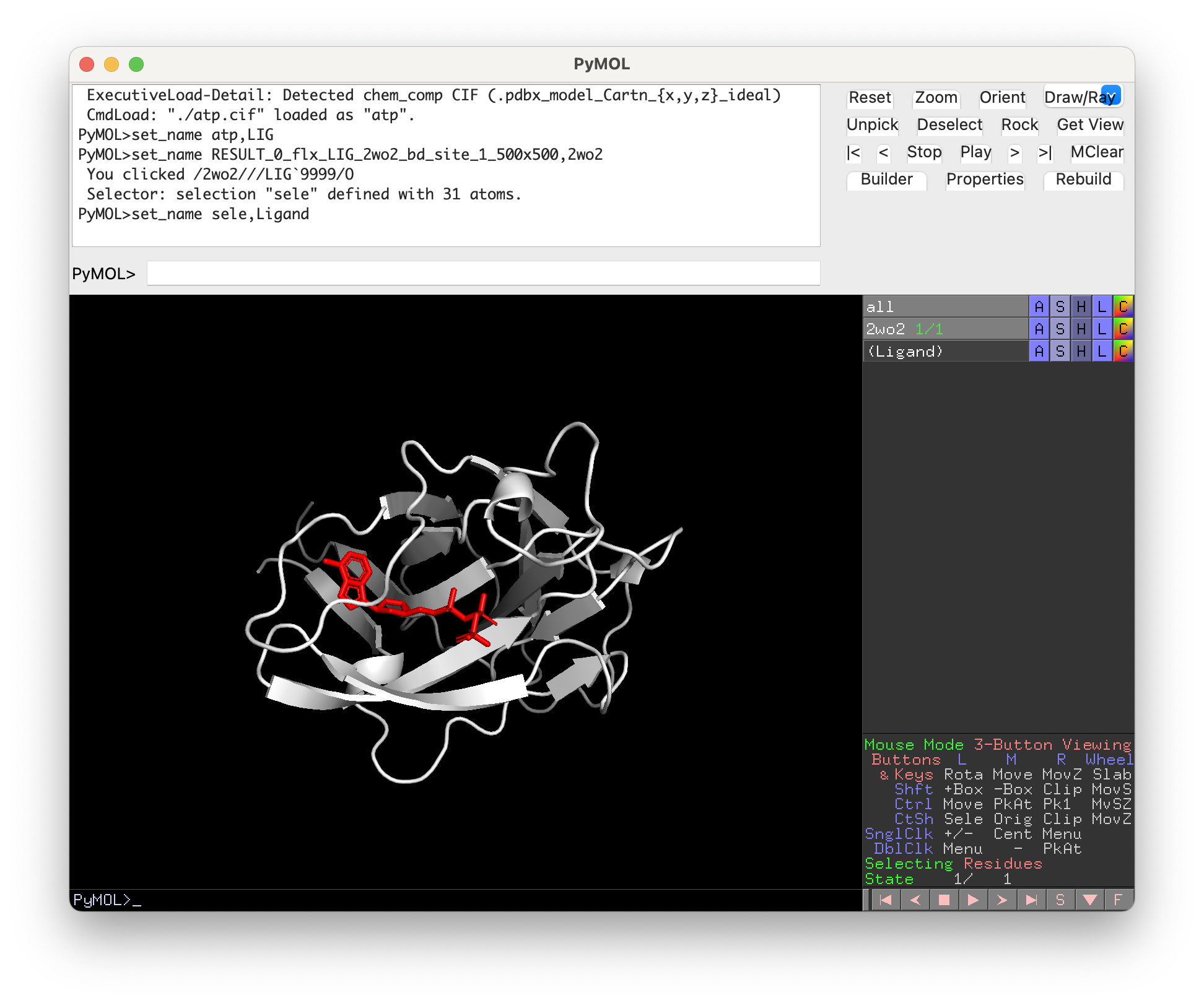
The visual output of Surfaces will be shown in the PyMOL window.
To visualize specific interactions click on the residue name and a selection called “sele_surfaces”, containing the residue will be crated and the residue will be zoomed-in and shown in lines representation.
The user can also create a selection containing “ALL” or the “TOP N” residues in the interface by pressing the “Interface” button. A selection called “all_residues” will be crated
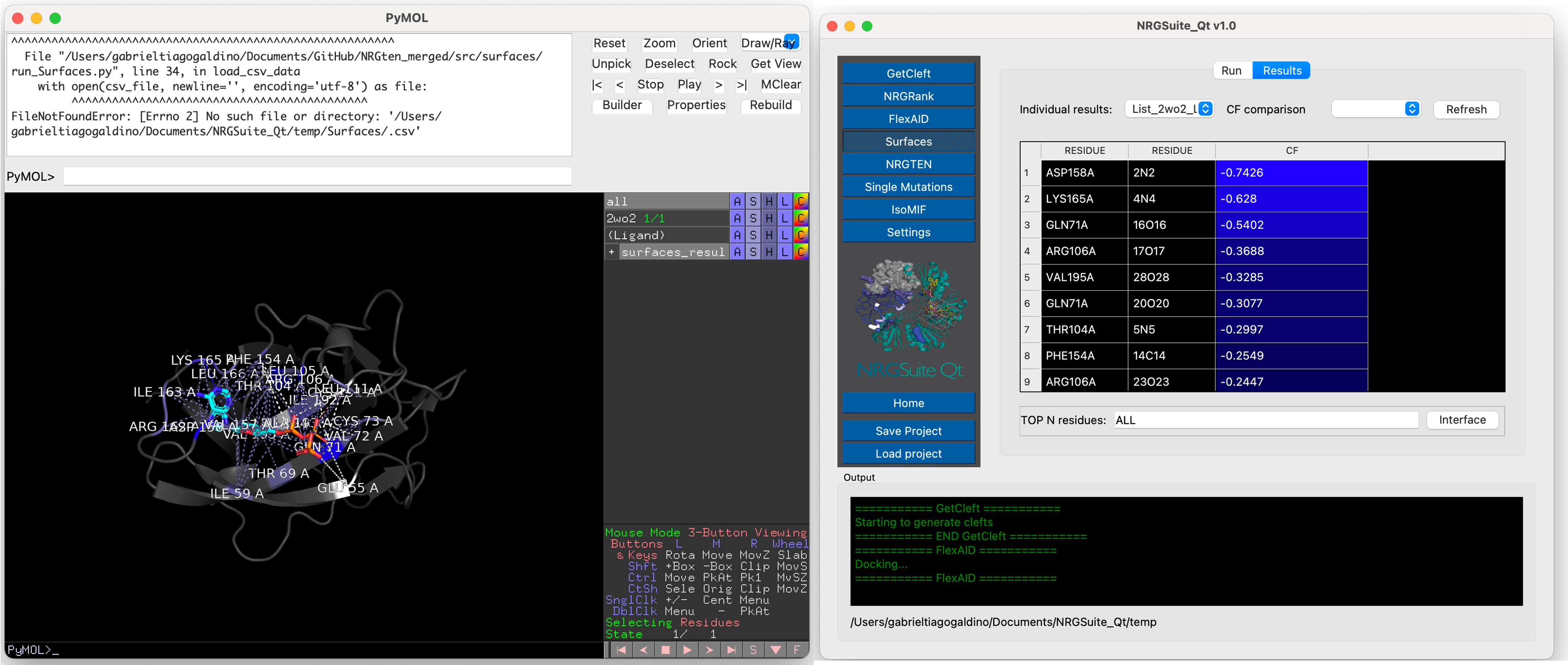
A list of all previous individual results will be listed in the “Individual results” list. In the case where a state with mutants is specified in the settings, the results of all individual mutants will be shown in the “Individual results” list. Press “Refresh” and the comparison of differences in total “CF” of each mutant in comparison with the wild-type will appear. All previous comparisons will be listed in the “CF comparison” list. All results are shown in the PyMOL interface in a group called “results_surfaces”